Wednesday, July 11, 2012
You can't ban redheaded sperm
It seems quite a few students have planned their high school and unversity careers in the hope that studying biology meant that could leave maths behind. So, when they are confronted with "p2 + 2pq + q2 = 1" and asked to do something with it, they are unhappy.
That particular formula is for something called the Hardy-Weinberg equilibrium and a significant proportion of students roll their eyes and slump their shoulders when you tell them they are going to need to use it for a problem. They think its arbitrary and irrelevant to anything the least bit important, and what's more it looks a little like it's already solved. So, I'm always looking for ways to convince people that Hardy-Weingberg isn't just simple, but actually intuitive and important. So here's my attempt to explain why knowing a little population genetics is helpful.
You may remember last year a Danish sperm bank had started turning away would-be donors with red hair, since there is little demand for sperm that might contribute to the creation of a readhead. It turns out, if you know a little bit about population genetics you can see that policy will have little effect on the number of red heads the sperm bank helps to bring into the world.
Hair colour is partially controlled by a gene called MC1R. There are different versions of MC1R floating around in human populations, and one of them has a mutation that stops melanin (a dark pigment) passing into hairs as they grow. Geneticists call different versions of a gene "alleles", so we'll call this flavour of MC1R the "red hair allele" and give it the symbol r.As I'm sure you know, you have two copies of most of your genes, one inherited from your mother and the other from your father. Red hair is a recessive trait, which means in order to have red hair both of your copies of MC1R need to be the r allele: if you have one or two copies of the "normal" MC1R allele (which we'll call "R") you have some pigment passing into your hair and it will be another colour. We call the total genetic make-up of a person their "genotype", and their physical characteristics their "phenotype", so here's a table showing the genotypes and the phenotypes we're talking about in this post:
| Genotype |
r/r
|
r/R
|
R/R
|
Phenotype (hair colour)
|
Red Hair
| Not Red Hair | Not Red Hair |
I know there are a lot of technical terms there (Carl Zimmer will not be happy...), but we do need to be precise when we talk about genetics because, strange as it may seem, there isn't a single definition of the word gene. Once you've got your head around the terms, it's all pretty straight forward: you need two copies of the r allele to have red hair. Think what this means for the Danish sperm bank though. Turning away red headed sperm donors doesn't turn away red headed sperm since there will still be "carriers" with only one copy of r (and, thus, non-red hair) donating sperm and half of those sperm will be "red headed sperm".
How big a problem is this likely to be? First we need to work how common the r allele is, and we can use the frequency of redheads to find that. By long tradition, the frequency of a recessive allele is denoted by "q", so, in a population where one quarter of the alleles are r we'd say q = 0.25. We know that in order to have red hair you need both your copies of the MC1R gene to be the r allele and that you inherit each allele separately. When probabilities are independant we can mutiply them, so the chace someone in this population is a redhead is q x q = q2 = 0.06 .Following the same logic, the frequency of the R/R genotype must be the frequency of R squared (by convention, the frequency of a dominant allele is called "p", so that's p2).
Knowing this relationship, we can work backwards and find the frequency of r if we know the proportion of redheads in a population. In most of Northern Europe, about 4% (0.04) of the populaiton are redheads so q2 = 0.04 and q = √0.04 = 0.2. As you can see, red hair genes can be a lot more common than redheads:


It's easy to calculate how the policy would work in populations with more or less redheads:

A lot of rare diseases are caused by recessive alleles. They remain rare for the obvious reason that people with such diseases are unlikely to pass on their genes. But they remain present in populations because, as we've found, once recessive alleles get rare the overwhelming majority of them are found in carriers. In this way, rare recessive alleles are seldom exposed to selection so they stick around for a long time.*
Because disease causing genes stick around in populations, there is a pretty good chance that you carry a few alleles that would cause a debilitating disease in someone who had two copies of them. The same applies to anyone you might be hoping to have children with. Thankfully, its very unlikely that your prospective mate with have disease-causing alleles for the same genes that you do. That is, as long as you look beyond the family tree when you look for a mate. If you have a child with someone who is closely related to you, you will have each inherited some of your genes from the same source, which increases the chances you share disease alleles.
*In fact, they often reach a point called "mutation-selection balance" in which the frequency of the allele remains static because new mutations re-create the allele as quickly as selection removes it. JBS Haldane was the first person to notice this, and he used his theory to create a very accurate estimate of the human mutation rate well before we knew what genes were made of!
Labels: genetics, population genetics, sci-blogs, science, teaching
Friday, May 20, 2011
3 tweets
Labels: evolution, genetics, sci-blogs, science, science communication, speciation, species, twitter
Tuesday, June 8, 2010
If some of us have Neanderthal genes, are Neanderthals us?
 I got a little bit starry eyed writing about the Neanderthal genome the other day. I chose to retrace the arc of scientific progress that links the initial description of Neanderthal man as something different than modern humans to the point reached last month, where we are able to tag some of those differences to a single gene. Most of the news stories about the Neanderthal genome focused not on the genes that made us different from them, but a small percentage of the genome that reinforced the continuity been them and us. Genetic evidence that Neanderthals interbred with the ancestors of some modern humans. The revelation of these ancient assignations has caused some quite sensible people to say some quite silly things about what species are and what Neanderthals were. So, perhaps I can compliment my slightly hazy earlier piece with a more hardheaded take on why Neanderthals remain a species unto themselves.
I got a little bit starry eyed writing about the Neanderthal genome the other day. I chose to retrace the arc of scientific progress that links the initial description of Neanderthal man as something different than modern humans to the point reached last month, where we are able to tag some of those differences to a single gene. Most of the news stories about the Neanderthal genome focused not on the genes that made us different from them, but a small percentage of the genome that reinforced the continuity been them and us. Genetic evidence that Neanderthals interbred with the ancestors of some modern humans. The revelation of these ancient assignations has caused some quite sensible people to say some quite silly things about what species are and what Neanderthals were. So, perhaps I can compliment my slightly hazy earlier piece with a more hardheaded take on why Neanderthals remain a species unto themselves.
Let's start with the evidence that Neanderthals interbred with the ancestors of modern humans. Modern humans (Homo sapiens) arose in Africa about two hundred thousand years ago, all modern human populations outside of Africa descend from a relatively small number of migrants who left that continent between eighty and fifty thousand years ago. When those migrants first left Africa and entered the Middle East they would have met other humans. The ancestors of the Neanderthal had moved out of Africa and established themselves in Europe and Central Asia thousands of years before. Until now we haven't known which of the four 'F's (fighting, fleeing, feeding or reproduction) followed that first contact, the Neanderthal genome has given us a clue.
When you compare individual DNA bases that are variable within modern human genomes to the corresponding sequences in the Neanderthal genome you find that non-African sequences match the Neanderthal sequence slightly (but significantly) more often than African sequences do. It's possible that this pattern is an artifact of our poor sampling of African genomic diversity (that observant nerd Christie does a good job of explaining how here) but for the sake of argument let's take it for granted that his pattern is the result of ancient interbreeding. The authors of the paper describing the Neanderthal genome estimate people with no recent African ancestry inherited between one and four percent of their genome from Neanderthals. That number is the same for Papuan and East Asian populations as it is for Europeans despite Neanderthals having lived alongside Europeans for thousands of years, suggesting any interbreeding that contributed to modern human genomes was limited to that first period of contact.
This is where the problems start. Having heard the news that Neanderthals and some of our ancestors might have once swapped genes some people remember that nice easy test of species-status from high-school biology. Something like "if two animals can interbreed then they're part the same species." So, are we Neanderthals; or are Neanderthals us? No. In fact, the Neanderthal genome serves to highlight some the mistakes we commonly make when start trying to define species.
Biologists have spent a lot of time arguing about just what a species is and how can delimit species from the creatures that we study, too often we've forgotten that those are two different arguments. DeLene from Wild Muse has a thoughtful overview of some of the factors that contribute to the "species problem" in her review of Jody Hey's book on the same topic. You should read her piece because the species problem really is a fascinating philosophical question, but I think most of the fights that erupt around competing definitions of species come from a failure to understand that defining species and organising critters into species are two different tasks. We've been studying speciation, the process by which new species arise, for a while now and we've developed a pretty good idea of how it works. Two populations stop interbreeding with each other, during that period of "reproductive isolation" genetic changes in one population can't effect the other so natural selection and random changes (called genetic drift) change each population independently. Species are populations which are on independent evolutionary trajectories.
Reproductive isolation drives the independence that is at the heart of what species are, but it's not the sine qua non of a species. James Mallet from University College London has made a special study of hybridisation, and he reckons 10% of animal species and a whopping 25% of plants interbreed with other species from time to time. As molecular tools have been applied to non-model organisms it's become increasingly clear that the "species barrier" is more porous than we'd thought, and species can maintain their independence even in the face of the occasional injection of genes from other species.(If you're interested in the wider question, I've written a bit on the species problem here. The short version is we should see competing "species concepts" as operational tools that might be used to help delimit species, but not as definitions).
Now, think about the results from Neanderthal genome. Most sequences in that genome are separated from their human counterpart by a split that happened over five hundred thousand years ago. There is pretty good evidence that Neanderthals and the ancestors of non-Africans interbred when they met each other in the Middle East about four hundred and fifty thousand years after that initial split. That gene flow had the potential to homogenise the two populations into one, but it didn't. Each lineage maintained its identity. For the twenty or so thousand years that Neanderthals continued to exist they retained identifiable morphological traits. There are fossils in Europe that some argue show a mixture of characters, but any interbreeding in that continent left no mark on modern European genomes, which have no more Neanderthal DNA than Papuan and Chinese genomes do. At the same time, the authors didn't detect any flow of modern human genes into Neanderthal genomes (so it's not a case of of modern humans swamping Neanderthal populations and erasing any trace of genetic admixture in the process). The available evidence seems to point o Neanderthals and modern humans as separately evolving populations, and a little bit of gene flow between them wasn't enough to upset that pattern.
I should stress, by saying H. neanderthalensis and H. sapiens are different species we aren't saying very much about how different Neanderthals were from us. Species are not defined by a degree of difference, or an essence that was missing in Neanderthals but is present in us, they're just another human population that was moving in a different direction (and eventually extinction). If some of us do have Neanderthal genes, then it only goes to show how fuzzy the line between our species and the rest of the biological world is.
Green RE, and many, many others (2010). A draft sequence of the Neandertal genome. Science (New York, N.Y.), 328 (5979), 710-22 PMID: 20448178
James Mallet's bit on the frequency of hybridisation is taken form here: Mallet, J. (2005). Hybridization as an invasion of the genome Trends in Ecology & Evolution, 20 (5), 229-237 DOI: 10.1016/j.tree.2005.02.010
The ideas about species and species delimitation presented above are pretty similar to Kevin de Quieroz's take:
De Queiroz, K. (2007). Species Concepts and Species Delimitation Systematic Biology, 56 (6), 879-886 DOI: 10.1080/10635150701701083
Labels: evolution, genetics, genomics, Human evolution, human genome, might interest someone, neanderthals, research blogging, sci-blogs, science
Friday, May 28, 2010
Living up to our name
Homo sapiens means "wise man". Sometimes it's hard to think that Linnaeus was right in honouring our species with that name. We're the reason the earth is going through its sixth great extinction; people are still routinely killed for belonging the wrong race, religion or sexuality and the prospect of taking action on climate change makes a significant proportion of the population behave like children. So it's nice to be reminded every now and again about the sorts things our species can do when we put our minds to it. I've been trying to find time to write a proper post about the Neanderthal genome, but here's something to think about on a rainy Friday afternoon.
In 1857 an anatomist and a school teacher, Hermann Schaffhausen and Johann Fuhlrott, described a set of bones that had been discovered in a limestone quarry in what was then called the neanderthal region of Germany. Amazingly, the neanderthal region was named after Joachim Neander whose own name translates as "new man". A new man was exactly what Schaffhausen and Fuhlrott saw in the bones that they described. They were at once human and something "other" Chief among the characters that set the neanderthal samples apart from modern humans was the thick brow ridge that we now think of as characteristic of primitive humans. Thanks to these differences the school teacher and the anatomist concluded that the neanderthal samples were human but something quite different than modern Europeans.

"Neanderthal Man" was the first pre-human fossil to be described. At the time science had no convincing mechanism by which species might change over time and no idea of how organisms passed on traits to their offspring. Within in a couple of years Darwin had published The Origin and Mendel his Experiments on Plant Hybridization (which was promptly ignored, and only cited three times in 30 years). In time scientists discovered more human fossils; Neanderthal man showed up all over Europe and took the name H. neanderthalensis, Euguen Dobois uncovered H. erectus in Asia and host of anthropologists have since added characters like the Turkana boy, H. habilis, Ardi and a whole cast of Australopithecines to our family tree.
The science of heredity moved on too. In the 20th century geneticists, especially Hugo deVries, rediscovered Mendel's work and set about building a particulate theory of inheritance. TH Morgan showed that genes resided on chromosomes, Fisher, Wright Haldane and others synthesized Mendelian genetics with Darwin's ideas on evolution, MacLeod and McCarty showed that DNA (a chemical initially identified by Miescher) contained genetic information (though no one believed them until Hershey and Chase demonstrated it again) and, of course, Watson and Crick showed us what DNA looked like thanks to Rosalind Franklin's x-rays. It took a little over 20 years to get from Watson and Crick's double helix to first complete virus genome and another 30 to scale the 5 orders of magnitude in size between that one and the human genome.
Last month, scientists published a first draft of the Neanderthal genome. 60% of the genetic make up of species of human that has been extinct for thirty thousand years. Thanks to the work of all those scientists listed above, and countless others who go unremembered, we now have a pretty good idea about the genetic basis of the thick brow ridge that convinved Schaffhausen and Fuhlrott than neanderthal man something different than other humans. The Runx2 gene is in a region of the genome that has been selected for in the H. sapiens lineage. We know from the work of yet more scientists that Runx2 is one of the most important genes regulating bone growth in humans and is associated with malformations of the skull. It's no great stretch to imagine that our species lost the brow ridge that that we associate with primitive humans thanks to changes in the expression pattern of Runx2.
It some ways that's a trivial piece of information, we've known for a long time that most morphological change is likely due to changes in the expression pattern of development genes. But isn't it wonderful to think that in the span of two human lifetimes we've moved from knowing nothing of our species' history to the point that we are developing hypothesis on the molecular basis of the changes that made us different from the host of human species we've since discovered.

Labels: evolution, genetics, genomics, Human evolution, human genome, sci-blogs, science
Sunday, April 4, 2010
Sunday Spinelessness - A Nobel Prize Winning Insect
I think invertebrates are important. The 95% of animal species that don’t have a backbone are not simply the base of the animal kingdom’s pyramid, they are the little creatures that run the world. A third of the planet’s food production relies on honey bees, collembola and corpse-feeding insects turn dead tissue into living tissue and coral reefs can turn the nutrient-poor tropical seas into submarine rainforests. There are even a couple of invertebrate animals that have won the Nobel Prize.

Drosophila melanogaster has probably taught us more about genetics than any other animal on earth. In the wild D. melanogaster larvae develop on rotting fruit so, just like the flesh-flies that were featured here a couple of weeks ago, they are faced with the problem of having to complete their entire developmental program in the short period of time the fruit they are born in is a viable food source. Thanks to these environmental constraints, D. melanogaster has a very short life cycle. Under optimal conditions they can go from egg to adult in a week. This remarkable developmental haste means drosophilists can run genetic experiments that cover many generations in a few months, and they can run many replicates of these experiments because each of them takes up about this much space:

Drosophila has been kept in laboratories since the the turn of the 20th Century but T.H. Morgan was the first person to put Drosophila at the forefront of genetic research. Morgan was an embryologist by training and, like a lot of embryologists then and now, he became interested in a school of evolutionary thought called mutationism. As the name suggests, the mutationists argued that one-off mutations were the creative engine of evolution, relegating natural selection to weeding out maladaptive mutants. In order to test the creative power of mutation Morgan grew up generation after generation of Drosophila and bombarded them with anything he thought might mutate them; radium, salts, sugars, acids, bases and even centrifugal force. Two years of this mutational bombardment got Morgan nowhere, he could induce changes in his flies but none that would be stably passed on. In 1910 he found a single white eyed male.
There is a story, which I can't find repeated by reliable sources, that holds that Morgan took the first white eyed male home with him in jar and slept with the jar next to his bed that night. I don't know if that story is true but that one fly does have a treasured place in the history of genetics. By crossing it to normal eyed (what geneticists call "wild type") females he was able to show that the genetic factor that made the fly's eyes white was part of the sex determining chromosome. For the first time a gene had been shown to be reside on a chromosome. A few years later he showed that multiple genes are arranged in linear fashion along chromosomes by demonstrating crossing over between the white eye gene and another called rudimentary. At Otago second year geneticists repeat Morgan's experiments, so this picture, sorting flies under a binocular microscope, will be familiar to anyone whose been through the program. (it will probably also bring back memories of escaped flies and a whiff of the (dilute) ether used to knock the files out...)

Morgan was awarded the Nobel Prize in 1933, in 1948 Drosophila research got another Nobel, this time to Hermam Muller for showing X-ray radiation could induce mutations. Geneticists have continued to use Drosophila as a model organism, perhaps most usefully in untangling the genetic interactions that underly the development process. In 1980 Christiane Nüsslein-Volhard and Eric Wieschaus presented the results of a mutational screen; that is, they mutated Drosophila stocks at random and recorded the developmental phenotypes that resulted. Nüsslein-Volhard and Wieschaus identified 15 genes involved with the very early stages of development. In quick time Drosophilists mapped that those genes to chromosomes and worked out how their products combined to pattern a developing embryo. Nüsslein-Volhard and Wieschaus' work laid the ground work for one of the most staggering findings of modern biology, almost all the genes that help shape the Drosophila embryo have counterparts in the human genome that play similar roles in our development. An insect can be a useful model for human development and disease genetics. Nüsslein-Volhard and Wieschaus were awarded the Nobel Prize in 1995, the third Nobel for work on Drosophila.
A big thanks to Sarah Morgan, one of Otago's fly pushers, for the photos that illustrate this post. Sarah's off to the US of A this week to show off her research at The Big Drosophila Meeting in Washington DC so she will probably have some less historical Drosophila science to talk about in the next little while...
Nüsslein-Volhard C, & Wieschaus E. (1980) Mutations affecting segment number and polarity in Drosophila. Nature, 287(5785), 795-801. PMID: 6776413
Rubin GM, & Lewis EB. (2000) A brief history of Drosophila's contributions to genome research. Science, 287(5461), 2216-8. PMID: 10731135
Labels: diptera, drosophila, environment and ecology, genetics, might interest someone, photos, sci-blogs, sunday spinelessness
Friday, March 26, 2010
Does a forty thousand year old finger point to another human species?
Here's the big figure from the paper, which was presented by Johannes Krause and colleagues in Nature yesterday. It's a phylogenetic tree which relates the little finger's mtDNA to H. sapiens and H. neanderthalensis sequences (click to see a high-resolution version):
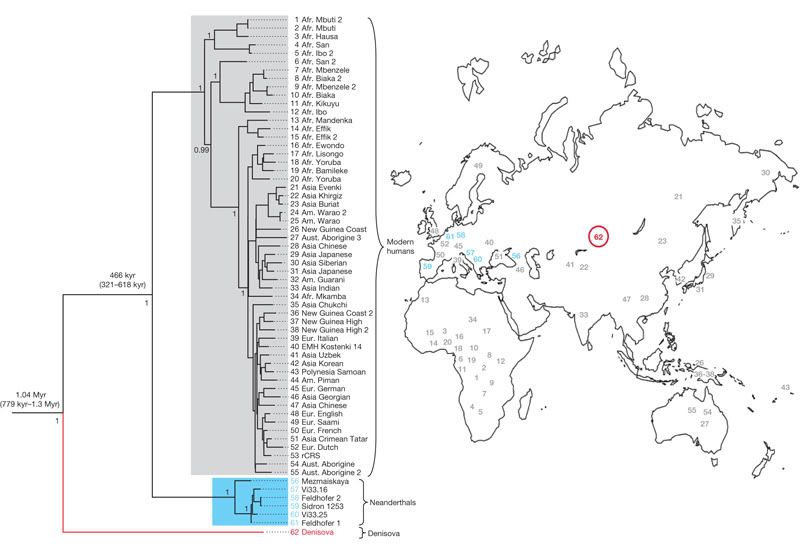
The Denisnova sequence is red, Neanderthal sequences are in blue and modern humans are grey. So, the Denisova mtDNA forms a distinct lineage that isn't represented in modern humans or in previously published Neanderthal sequences. By using the tree as the basis for molecular dating the researchers were able to estimate that Denisova lineage separated from other human mitochondrial lineages between 0.78 and 1.3 million years ago. The temporal context the molecular dating adds to the phylogenetic tree helps to us understand where this new mitochondrial lineage might fit into humanity's family tree.
I've said before that most of our species' history was played out in Africa, and, in fact, the same is true when we step up a taxonomic level and look at our genus. All the human species that have been found outside of Africa descend from migrants that moved out of that continent at some stage. Here's a schematic representing some of the species in the wider human family tree and the timing of the migrations that moved them out of Africa.
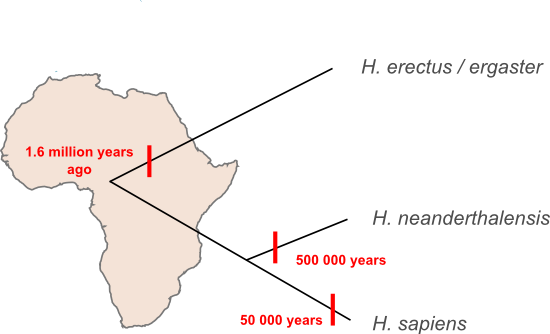
How does the new evidence presented by Krausse et al. fit into that scheme? Perhaps the simplest interpretation is the the Denisova lineage represents a new species. The estimated age of the Denisova lineage makes it too young to have been carried out of Africa by the first wave of H. erectus migrants to leave Africa and apparently too old to have been inherited from the migrants that went on to form the Neanderthal lineage. If the Denisova sequence is something new then we'll have to update our family tree, adding a new branch and a fourth migration out of Africa.
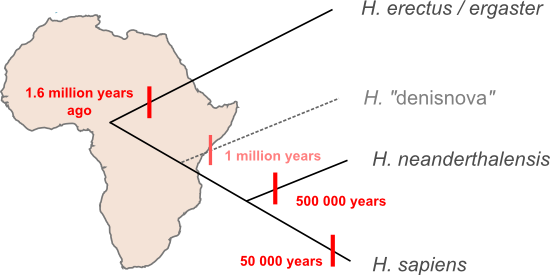
John Hawks thinks we should hold off on updating the family tree too qucikly. The Desinova specimen might be a Neanderthal. At first glance the tree presented by Krausse et al. seems to dispel that possibility since previously identified Neanderthal sequences are more closely related to modern human sequences than the new linaeage, but that tree is based entirely on mtDNA. The mitochondrial genome is inherited as if it was a single gene. We can often use trees estimated from a single gene ("gene trees") as a proxy for species-level relationships ("species trees") but, in fact, every gene in a population has its own history and there there are scenarios that can push a given gene tree away from underlying species tree. Perhaps the easiest way to visualise how you'd end up with mitochondrial lineages that diverged millions of years ago within a single species is to think about genetic lineages moving through a population while speciation happens. New species form when populations stop sharing genes with each other, in the diagram below the big black triangle represents a barrier to gene flow. What happens if multiple different gene lineages are present in the ancestral population at the time that this gene flow stops? Usually, given enough time, each species will "sort" into specific gene lineages that descend from just one of the lineages in the ancestral population, but it's also possible for one (or both) species to maintain multiple lineages for some time. Such "incomplete lineage sorting" makes gene trees bad proxies for species trees and it's just possible that something like this has happened in Neanderthals:
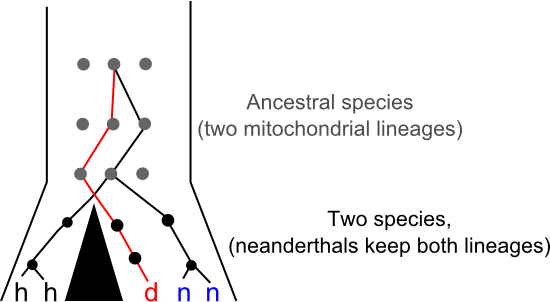
Perhaps by moving to the very Easterm edge of the Neanderthals range we've sampled for the first time a lineage that existed in that species for the whole time it was in Europe. Maybe, and Hawks surely knows a lot more about paleobiology than I do, but I don't really buy it. It's certainly possible for a species to harbour deeply divergent mitochondrial lineages, but the time it takes for gene-lineages to sort within a species is relative to the effect population size of that species. Neanderthals probably had a relatively small effective population size (and mtDNA definitely does, since only females pass it on and then in only one copy) making the retention of multiple lineages over hundreds of thousands of years seem like a long shot. As Hawkes argues, strong geographic structure in Neanderthal populations might have aided the retention of divergent genetic lineages against those odds, maybe the Denisova mitochondrial lineage was extinct in Western Europe but common in Central Asia? It's possible, but I wouldn't bet on it.
Finally, the Denisova sample might be our first look at H. erectus DNA. H. erectus remains have been recovered from China so it seems possible they were in Siberia too. As I've said, the molecular dating of the Denisova lineage probably makes it too young to be a descendant of the first wave of migration form Afirca (though, of course, there is some uncertainty associated with that dating), but it might be evidence of genetic exchange between African and the H. erectus diaspora. As we've come to understand the origin of our species we've realised that the simple "Out of Africa" model is just that, a model, and the true pattern is more complex. H. sapiens really did have its start in Africa and it really did push out into the rest of the world in the last 50 000 years or so, but during that expansion populations have continued to exchange genes. There's no reason to believe that that H. erectus could not have done the same, perhaps the main thrust of the H. erectus expansion was 1.6-2 million years ago but genes continued to flow in and out of Africa for sometime after that.
So, there are three possibilities for the Denisova sample:
- It could be a new species,
- It could be an ancient mitochondrial lineage retained in eastern Neanderthal populations but lost elsewhere
- It could be the first H. erectus sequence.
Krause, J., Fu, Q., Good, J., Viola, B., Shunkov, M., Derevianko, A., & Pääbo, S. (2010). The complete mitochondrial DNA genome of an unknown hominin from southern Siberia Nature DOI: 10.1038/nature08976
Labels: evolution, genetics, Human evolution, might interest someone, phylogenetics, sci-blogs, science, speciation
Friday, February 26, 2010
Nucleotide diversity - what two new African genomes mean
As I've said before there really is no such thing as the human genome. There are millions of differences between individual genomes and we are each born with about 150 new muations. In an age in which we can sequence assemble and analyse entire genomes in two years understanding the breadth of human genetic diversity is at last an achievable goal and if you want to understand human diversity then you need to look to where we came from. Trace any family tree back far enough and you will end up in Africa and, in fact, most of human history was played out entirely in that continent. Modern humans arose in Africa about 250 000 years ago and only spread out to Europe and the rest of the world in the last 60 000 years, displacing Homo erectus in the process. The migrants that founded the modern European, Asian and American populations would have carried with them only fraction of humanity's genetic diversity when they left Africa but untill recently genomics has focused on those populations. Until last week the two African genome sequences available to researchers were both from Yoruban volunteers to the hapmap project. Although those sequences are very useful they represent only one tip in the deeply branching tree of humanity

To broaden our understanding of African genomes Schuster et al looked to the South of the continent and at two people in particular. !Gubi is a Khoisan (or bushman), a member of a one of the earliest diverging groups within the humanity while Desmond Tutu hails for various Bantu peoples. The results taken from theses genomes along with lower density sequencing and genotyping of other Bantu and Khoisan volunteers reinforces just how much genetic diversity exists within Afirca. By using a method called principle component analysis to reduce a the correlations among millions of single base pair differences (single nucleotide polymorphisms or SNPs) to a smaller set of uncorrelated vectors you can see patterns in the genetic diversity of groups. Applying this method to West African (Bantu and Yoruba), Khoisan and European populations reveals the comparative genetic homogeneity within Europeans and that the difference between the two African groups is comparable to that between either of them an Europeans.
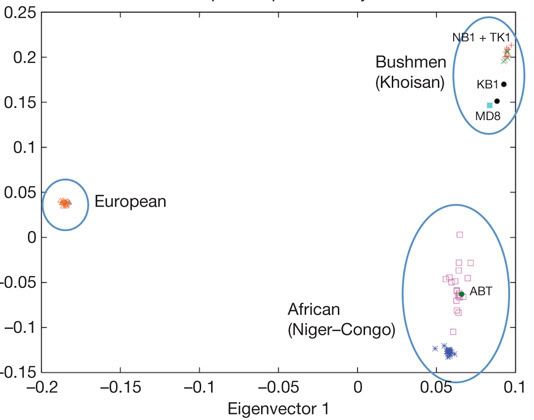
The paper is available to under a creative commons license here and if you feel suitably qualified you can play with their data which has been released on the Galaxy framework.
Labels: evolution, genetics, genomics, Human evolution, human genome, research blogging, sci-blogs, science
Friday, January 15, 2010
The why of the Y-Chromosome's amazing evolutionary rate

There is something faintly pathetic about the Y-chromosome when its lined up with its peers in a karyotype. Each of the 22 numbered chromosomes pair off with a near identical partner just their size while the Y has to shape up to the X which has more than twice as much DNA and 25 times as many functional genes.
The puny Y-chromosome only looks worse when you realise that mammalian sex chromosomes weren't always so mismatched. 160 million years ago the X and Y were just another pair of chromosomes, albeit the pair that the carried the sex determining gene SRY. Over time the chromosome that went on to become the Y stopped swapping genes with its partner, allowing it to maintain a suite of genes that are beneficial in male bodies but not in females. It's the lack of genetic recombination that sent the Y into its decline. Genes on any other chromosome can be swaped between pairs, meaning over many generations individual gene copies (called alleles) are exposed to natural selection independently of alleles either side of them. The same process doesn't apply to alleles on the Y-chromosome. Since the Y is always passed on as a single unit natural selection acts on the whole thing - a broken gene might make it into the next generation because it is attached to beneficial mutations. The efficiency of natural selection is further reduced in the Y-chromosome because it has a relatively small effective population size (less that one quarter of that for normal chromosomes since only males carry the Y and then in only one copy and even then a larger number of males than females don't contribute to the next generation) which makes genetic drift a strong force.
What we've known about the Y-chromosome's past has has shaped out ideas about what it is now and what it will become. Until quite recently the Y was seen as more or less a derelict chromosome, a few broken remnants of the genes still found on the X and a couple of male-specific genes hanging on the the sex determining gene SRY. People have even go so far as to extrapolate the Y's long slow decline to a future time at which the Y will simply disappear. The first clue that the Y-chromosome might be a little more resilient than that came in 2003. The publication of the complete sequence of the human Y-chromosome revealed more than fossils from the Y's more substantial ancestor. There are plenty of those so called "X-degenerate" segments but most of the active genes in the Y are in large repetitive runs of DNA called the "ampliconic regions". The genes in these regions are mainly made of DNA sequences unique to the Y chromosome and are expressed only in the testes - suggesting the Y has been making its own genes at the same time that its been losing the X-degenerate ones.
Untill this week it has been hard to test the idea of a regenerating Y-chromosome in an evolutionary framework. Those large repeated runs of DNA are very hard to sequence (the standard metaphor is putting together a jigsaw puzzle made entirely of sky) so we haven't had another Y-chromosome sequence to compare ours with. Now, thanks to Jeniffer Hughes and colleagues, we do and the result it stunning. Not only has the Y-chromosome been making genes, it's been making them at an outrageous rate. Thirty percent of our Y-chromosome sequences have no counterpart in the chimpanzee. As the authors say that's the sort of divergence you'd expect to see between humans and chickens, which are separated by 310 million years of evolution not humans and chimps which only split 6 million years ago!
It's evident that, far from being in the tail end of an inexorable decline, the Y-chromosome is evolving a good deal more quickly than the rest of the genome. So, the burning question is what is behind that evolutionary rate? There is probably no single answer to that question but it's safe to assume it results from some of the unique features of the Y-chromosome; a lack of genetic recombination, the presence of those large repetitive sections of DNA and the preponderance of male specific genes.
It's usually a good idea when trying to explain an evolutionary phenomenon to think of explanations that don't invoke natural selection as the main driver as a sort of null hypothesis against which to test other ideas. In this case the increased fixation of new genes on the Y-chromosome might simply reflect an increased rate of production of new genes. Those highly repetitive sections of the Y-chromosome are the perfect substrate for a process called ectopic gene conversion in which a Y-chromosome can recombine with itself and as a result duplicate streches of DNA. We know from human studies that a process like this has made wide scale structural changes in the last 100 000 years and it might be enough to explain the Y's unusual gene production.
I think it's very likely that natural selection also plays a role in the number of of those new genes that become fixed in the human and especially the chimp lineage. Most of the active genes on the Y-chromosome are expressed in the testes and involved in sperm production. Chimpanzees are highly polygynous polygynandrous [Thanks to Harvest Bird for pulling me up on this,], in most cases a female will mate with each of several dominant males in a troop, and a result sperm competition is an important level of selection. Although humans aren't as polygamous as chimps (and likely haven't been in our recent history) it's clear that fertility selection is still an important force and we know for sure that mutations in the Y-chromosome can lead to infertility so, again, the fate of new genes on the Y-chromosome are likely to be driven by selection.
Both the adaptive and non-adaptive explanations above might will be influenced by the lack of recombination in the Y-chromosome. The reduction in the efficiency of natural selection described above will stop very slightly deleterious mutations from being driven to extinction which might mean new genes that would be selected against on any other chromosome become fixed on the Y. This phenomenon can be enhanced when it is coupled with selection producing a 'selective sweep'. If a new beneficial mutation, perhaps associated with sperm competition or fertitily selection, pops up in on a chromosome with a bunch of other mutations that whole thing will be selected for and driven to fixation which has the potential to make for large scale changes quickly.
It is likely that the amazing evolutionary rate of the Y-chromosome is a result of some combination of all these factors but it should be possible to disentangle at least some of their contributions. If sperm competition is a major driver of Y-chromosome evolution then it follows that animals that go in for purely monogamous relationships will have comparatively low rates. Evolution has furnished us a natural experiment to test this idea, all gibbon species form pair bonds and are highly monogamous. We could test the sperm production hypothesis by sequencing the Y-chromosome of two gibbon species and calculating the rate of evolution of a Y-chromosome in a monogamous species. .Although I'm happy to present the test of this idea I'm not going to line up to do it, those repetitive sections of DNA make sequencing Y-chromosome so hard that it took 13 years to do the human one and 8 to finish the chimp one.
Hughes, J., Skaletsky, H., Pyntikova, T., Graves, T., van Daalen, S., Minx, P., Fulton, R., McGrath, S., Locke, D., Friedman, C., Trask, B., Mardis, E., Warren, W., Repping, S., Rozen, S., Wilson, R., & Page, D. (2010). Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content Nature DOI: 10.1038/nature08700
Labels: chimpanzee, evolution, genetics, genomics, Human evolution, might interest someone, sci-blogs, science, Y-chromosome
Thursday, October 15, 2009
Human genomes
When I started out as a genetics student the big goal everyone was talking about was understanding "The Genome" - that monolithic set of DNA bases that make us human. Of course, there is no such thing. Pick two human genomes at random and you'll likely find 2 million single-base differences and plenty of structural differences on top of that. As with just about everything in life understanding the range of variation and the diversity in human genomes is much more interesting than focusing on the average of that diversity.
The publication of a draft sequence from the Human Genome Project in 2001 really was the begining of a new epoch in genetics (and a revelation in itself- it takes 20 000 genes to make a nematode and 25 000 to make us?) but the real value of that project has been the generation of a scaffold that subsequent projects like the super-optimistic 1000 genomes project, hapmap and the genographic project (which New Zealand researchers contribute to) have been able to inherit in their attempts to understand genomic diversity. This week Nature has a special issue focussing how data built on information from the Human Genome Project is contributing to the way we understand disease and even allowing personalised testing for genetic risk.
Unfortunately they've stuck one of the most interesting articles behind a pay wall (Nature, the people that bring you a special on science and society but don't let society read it...) Bruce Lahn and Lanny Ebenstein have an opinion piece on genetics and race.
The current moral position is a sort of 'biological egalitarianism'. ... the view that no or almost no meaningful genetically based biological differences exist among human groups, with the exception of a few superficial traits such as skin colour. Proponents of this view seem to hope that, by promoting biological sameness, discrimination against groups or individuals will become groundless.Of course, as Ebenstein and Lahn point out, there is a problem with this view - race almost certainly does have a genetic basis beyond a few superficial traits. To take the local slant Polynesians and in particular Māori represent the furthest extent of the series of migrations that followed our ancestors moving out from Africa. Settling the Pacific must have involved a series of population bottlenecks - events in which small groups form a new population representing only a fraction of the genetic diversity in the parental population. Such bottlenecks will have inevitably left a mark on the gene pool of Māori and Pacific Island populations that more recent interbreeding won't yet have erased. There will be some genes that are unique to Polynesian populations and others that are orders of magnitude more or less common than they are in other populations. If we embrace that genetic diversity we might be able to understand why Māori face a much greater risk to, for instance, diabetes, gout and liver disease than Pakeha. To, as people have really suggested, ignore that genetic diversity because racists might use it to further their stupid cause is, in the words of Lahn and Ebenstein "llogical, even dangerous" Oh, and by the way, whatever we find out about the genetic basis of race I think it's safe to say that genes won't care too much for national borders - but that's not going to stop the UK from genetically screening assylum seekers...
Labels: genetics, genomics, human genome, sci-blogs, science and society
Tuesday, September 29, 2009
Where did you get that preposterous hypothesis
From time to time you find yourself disagreeing with something you read in a scientific paper. Perhaps you don't think the authors have applied a method correctly or ,more often, you don't think that the results they present are enough to justify the claims made in the their discussion or their university PR department's breathless press release. You don't often end up wondering if the third most prestigious journal in the world might have an April Fool's day issue. But what else is one to think when confronted with an opening paragraph like this one from a recent paper [1]:
I reject the Darwinian assumption that larvae and their adults evolved from a single common ancestor. Rather I posit that, in animals that metamorphose, the basic types of larvae originated as adults of different lineages, i.e., larvae were transferred when, through hybridization, their genomes were acquired by distantly related animals.
Got that? The author thinks that animals with distinctly different larval forms (caterpillars and butterflies, tadpoles and frogs, veligers and marine snails...) don't descend from a single ancestor that had a simple life history and later developed a two-stage strategy. Rather, Donald Williamson thinks that metamorphosing organisms are chimeras - hybrids between two distinct lineages in which the two parental genomes have reached a compromise such that one parent gets to run what we call the larval form and the other oversees the adult.
This is certainly not a mainstream idea, but the paper I'm talking about was published in the Proceedings on the National Academy of Science (PNAS), one of the most prestigious scientific journals that there is, Williamson must have some good data to support his idea right? Well, no. Williamson's entire case appears is that he finds it really really hard to imagine metamorphosis evolving in gradual steps and, besides, some larval forms look quite a lot some other organisms. Williamson does distinguish himself from other pedlars of what Richard Dawkins has named the "argument from personal incredulity" by at least providing a specific hypothesis to test: modern insects with 'caterpillar' larvae (butterflies, beetles, ants, wasps, bees, files...) descend from an 'accidental' mating between a flying insect and an onychophran (no illustration of this process is provided).
 Peripatoides novaezealandiae, a wide spread New Zealand endemic onychophoran and young, photo © Te Ara
Peripatoides novaezealandiae, a wide spread New Zealand endemic onychophoran and young, photo © Te Ara
Onychophorans (which we usually call 'peripatus' in New Zealand) are part of that admittedly large list of creatures that can be called "David's favourite animals" so before we hang Williamson's preposterous hypothesis out to dry I'm going to have to subject you to a little bit of cheer-leading. This is not the first time that onychophorans have been the subject of woolly evolutionary thinking. Since they are likely related to some of the most spectacular cambrian fossils people have called them "living fossils" and you'll even sometimes hear it proposed they represent a *shudder* "missing link" between arthropods (insects, crustaceans, spiders...) and annelids (earthworms and their kin). Which is all a great shame because it diverts attention from the fact the onychophorans are nocturnal hunters which crawl through the leaf litter on hydro-statically inflated legs in pursuit of small invertebrates which they immobilise with a sticky glue they spray from their mouths in order to let them inject digestive enzymes into their stricken prey and suck the resulting soup from its lifeless body. That's the sort of thing people ought to know about it.
What about Williamson's "larval transfer" idea? Is this a case, like Wegner and continental drift or Bretz on ice ages, in which science needs some outré thinking to get itself out of a rut that is holding it back? Hardly.
Insect metamorphisms isn't that hard a problem

An adult cicada emerging from its last nymphal molt © Te Ara
.Just how complete metamorphosis of the sort you see in butterflies evolved is a genuinely difficult and, as such, interesting question. But it's one that Williamson clearly hasn't bothered to read about. If he had he would've found a lovely review from Deniz Ereyilmaz2 who traces the history of the problem and makes a case that larvae are effectively free living embryos (an idea that was articulated by Harvey (of the circulatory system) and later used by Darwin in the 6th edition of The Origin to reply to contemporary criticism that his theory couldn't explain metamorphosis). Specifically, the idea is that the holometabolous insects (the ones that undergo complete metamorphosis) evolved from direct developing insects like cicadas and grasshoppers. In these insects the final stage of embryonic development is called a pronymph, in most species the pronymph molts into a mini-adult (called a nymph) before it hatches but a few species actually hatch as pronymphs. Ereyilmaz and the few entomologists that have tackled this question in recent years think holometabolous insects descend from species in which the pronymph hatched and then became able to feed. From there the development of the pronymph stage was extended while nymphal development (which usually proceeds as small changes accrued in each of several molts) was progressively squeezed into one step, which we now call pupation (like a caterpillar's cocoon).
There is some nice genetic evidence that something like that process has happened. One of the genes required to start the metamorphosis process is called broad, mutants that can't produce functional broad protein fail to pupate. Insects like cicadas and grasshopers that don't undergo complete metamorphosis also have copies of broad but in these insects broad is expressed at each nymphal molt - consistent with the idea pupation in holometabolic insects corresponds to nymphal molt in direct developing insects.
The evolution of complete metamorphosis remains an interesting question (if you are want to learn more Christopher Taylor has deeper look at it than I've given here) but the sort of path laid out above - the gradual addition of multiple, relatively small changes to the existing insect life cycle is surely orders of magnitude more likely than two genomes being thrown together and, somehow, deciding to regulate two complete separate developmental programmes as well as the entirely new process of breaking down the first genomes animal before development of the second one can begin?
Complete metamorphosis doesn't use two sets of genes
Williamson also asks 'genomocists' to search for distinct genomes within the DNA sequence of holometabolous insects. We don't need a complete genome to know that the same genes are being used in the development of adults and larvae. People have been studying the genetic basis of development in Drosophila (which my taxonomic pedantry won't allow me to call fruit-flies) for at least 20 years - all Williamson needed to do to check his hypothesis against the evidence was open an undergraduate textbook. Had he done that he would have seen, in one tirivial example, that the that patterning of the adult wing in Drosphila requires the genes hedgehog and wingless (geneticists usually name genes after what loss of function mutants look like ) both of which are also vital to defining the polarity of the segments formed in embryonic development. We've also know since at 1997 [3] than oncychophorans and insects inherited their hox genes, (the genes that lay out the basic body plan in animals) from a common ancestor that lived before the two groups split up - and the holometabolic insects we've looked at only have one set of hox genes.So why is this in PNAS?
OK, so Williamson has his answer to the problem of metamorphosis and no evidence is about to sway him from it. But he's not asking for his nonsense to be taught in public schools or anything - he's just a harmless crank. The question is why was his idea afforded space in one of the most prominent scientific journals instead of being expressed in the standard media for cranks - self published pamphlets or a huge single page website made with Mircrosoft FrontPage and featuring five different colours of text interspersed with clip-art and presented on a yellow background. Well, until very recently there where two ways to be published in PNAS, you could submit an article to the editorial office in the normal way or you could have a member of the National Academy of Sciences 'communicate' your article - which still required peer review but the whole process, including picking the reviewers, was overseen by the communicating member. In this case the communicating member was Lyn Margulis who richly deserves to be a member of the academy for providing the evidence then championing the very unorthodox idea that mitochondria (of which I've spoken before) descend from free living bacteria that long ago formalised a symbiotic union they'd fallen into with an ancestor of us Eukaryotes. That very strange idea has now been accepted by pretty much everyone that has an opinion on the matter but since that triumph Margulis has fallen into what Jerry Coyne( world famous geneticist, new atheist and cat blogger) calls Big Idea Syndrome. A lot of people who discover some interesting and important wrinkle in a prevailing theory get it into their minds that their discovery is actually driving force behind an entire field of study. In Margulis's case this syndrome manifests itself in an unwavering belief that all the interesting questions in biology can be answered with symbiosis and "acquired genomes" while modern evolutionary biology and its fascination with competition as a driver of change is "a minor twentieth-century religious sect within the sprawling religious persuasion of Anglo-Saxon Biology."
PNAS is ending the peculiar institution that saw Williamson's paper published, probably in part because papers appearing in the journal are treated with a degree of scepticism by at least some readers. I actually think that's a shame, the communicated papers had the potential to give a platform for important ideas that might otherwise be too unorthodox to appear in widely read journals - Margulis' original paper was rejected 7 times before the Journal of Theoretical Biology published it. It's a great pity a person whose work makes one of the best cases for the need for original thinking in science has has helped to highlight what happens when such creativity isn't met with a more critical mindset.
[comments are closed for obvious reasons, if you have something to say try the sciblogs version of this post] [1] Williamson, D. (2009). Caterpillars evolved from onychophorans by hybridogenesis Proceedings of the National Academy of Sciences DOI: 10.1073/pnas.0908357106 [2] Erezyilmaz, D. (2006). Imperfect eggs and oviform nymphs: a history of ideas about the origins of insect metamorphosis Integrative and Comparative Biology, 46 (6), 795-807 DOI: 10.1093/icb/icl033 [3]Grenier, J., Garber, T., Warren, R., Whitington, P., & Carroll, S. (1997). Evolution of the entire arthropod Hox gene set predated the origin and radiation of the onychophoran/arthropod clade Current Biology, 7 (8), 547-553 DOI: 10.1016/S0960-9822(06)00253-3
Labels: developmental biology, Evoution, genetics, might interest someone, sci-blogs
Monday, September 7, 2009
I TOLD you you're all mutants
Our typical conception of mutation is drawn from the tragic effects of those relatively rare mutations, induced in our bodies or passed on through germ cells, that lead to diseases (or, in movies to super powers). In fact, we are, each of us, mutants. DNA replication is not perfect, we are born with about 6 or 7 new mutations...
Well, a paper published last week[1] proved my general point while proving me wrong on the detail by a factor of 20 or so. A team of British and Chinese researchers that work with a family that has a unique Y-chromosome linked hearing disorder sequenced the entire sequence of the Y-chromosome from two men and found four mutations. Scaling up from the Y-chromosome to the whole genome then dividing by the combined 13 generations that separate the two men they arrived a mutation rate of 3 x 10-8 changes per nucleotide per generation. That would give us between one and two hundred new mutations.
This finding isn't actually a revelation. We had an idea of the rate of mutation in the human genome before we even knew what a gene was made of. JBS Haldane, one of the founders of evolutionary genetics and perhaps the only person to have enjoyed the First World War, used his theory of mutation selection balance to estimate new haemophilia causing mutations occur about once in every 105 generations.[2] When you consider that the gene responsible for Haemophilia A contains about 7 x 103 nucleotides and changes to many of those won't cause Haemophilia Haldane's estimate looks pretty good.
In fact, the Cool New Stuff in this paper isn't really the number that they've produced - that number is similar Haldane's esimate and to the measurble error rate of the enzymes that replicate our DNA and to the rate inferred by comparing our genome to that of the cimpanzee *. What's really neat is the fact they directly measured the rate by resequencing the whole Y-chromosome - that's more than 10 million bases to sequence, 35 at a time, and put together to check for mutations. The sort of project that would only have been possible as part dedicated genome sequencing projects a couple of years ago. With only two people and four mutations the estimate has wide error bars but it does pave the way to more accurate estimates for particular areas of the genome (including those underlying for diseases) and particular lineages of organisms (which is important for us evolutionary biologists)
I can't revel in my earlier post being confirmed in the broad sense without apologising for misleading you in the details. I was just flat out wrong when I claimed we all have 6 or 7 new mutations - I used a number that I had in my head and didn't bother to look it up. You can see where my number came from once you consider that only about 4% of the genome is functional DNA - 150 mutations in your genome will lead to about 6 mutations in functional regions. Still, the original is (about to be) modified and I am suitably shamed.
* As Larry Moran points out taken together these studies tell us something about the way evolution works. If the observed rate of mutation in DNA replication is not wildly different than the inferred rate of mutation in a pedigree or between closely related species most mutations aren't being selected against - more evidence for the importance of neutral theory in molecular evolution. back to the story ^
[1] Xue, Y., Wang, Q., Long, Q., Ng, B., Swerdlow, H., Burton, J., Skuce, C., Taylor, R., Abdellah, Z., & Zhao, Y. (2009). Human Y Chromosome Base-Substitution Mutation Rate Measured by Direct Sequencing in a Deep-Rooting Pedigree Current Biology DOI: 10.1016/j.cub.2009.07.032
[2]J. B. S. Haldane (1935). The rate of spontaneous mutation of a human gene Journal Of Genetics DOI: 10.1007/BF02717892
Labels: genetics, might interest someone, sci-blogs

All content not otherwise marked is licensed under a Creative Commons Attribution-ShareAlike 3.0 Unported License.